|
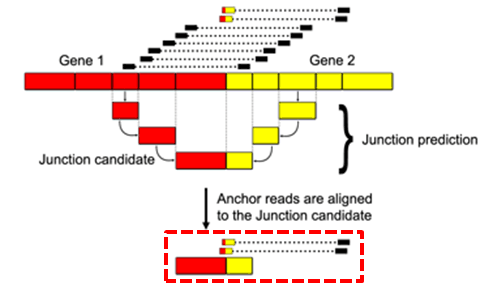
Each filtered Bridge event is scanned against the filtered Anchor dataset. If one of the two genes associated with a specific Bridge event corresponds to a mapped gene in the Anchor dataset, the matched unmapped read is aligned to the candidate Junction regions (red box) of the Bridge event using a dedicated built-in gapped alignment algorithm. The result of the alignment is then evaluated by a first, computationally fast, scoring algorithm. Alignments passing the first filter are evaluated by a second, more accurate, scoring algorithm. If the alignment succeeds, the Junction data is deemed to be valid (Junction read).
|